01
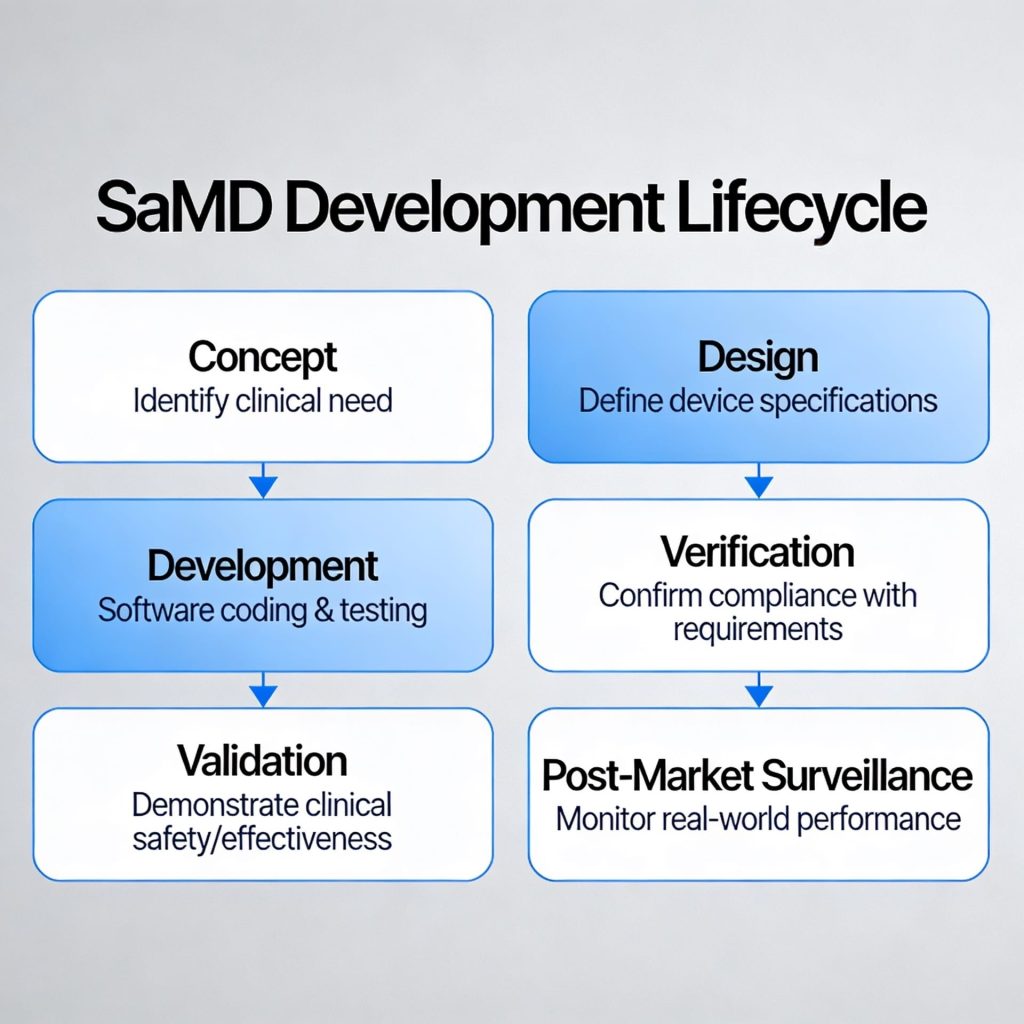
SaMD Engineering & Architecture
Mobile, cloud, and AI/ML engineering specifically built for SaMD, with architecture, documentation, and validation strategies aligned to the product's intended use, risk profile, and regulatory pathway.
- Native iOS and Android apps where the platform difference matters; cross-platform where it doesn’t
- Cloud-native architectures designed for clinical data volume, HIPAA-ready from day one
- Component architecture that lets a single codebase serve multiple product configurations
- Software Development Kits (SDKs) for interoperability with other devices in the clinical ecosystem