
A mobile medical app (MMA) is a software application running on a smartphone or tablet that performs a medical function — diagnosing, treating, monitoring, mitigating, or preventing disease. The FDA regulates MMAs that meet the definition of a medical device and carry significant patient risk; many lower-risk MMAs fall under enforcement discretion and are not actively regulated.
The five-step decision tree below walks through how to determine which category your app falls into — written by Sequenex’s SaMD development and regulatory specialists and updated for the 2026 FDA guidance.
Is Your Mobile Medical App FDA-Regulated? A 5-Step Decision Tree
Each step below asks one question. Your answer routes you either to the next step or to a final outcome — “likely subject to FDA regulation” or “likely under enforcement discretion.” Use this tree as a preliminary guide. Final FDA classification typically requires submission review or pre-submission consultation with the FDA.
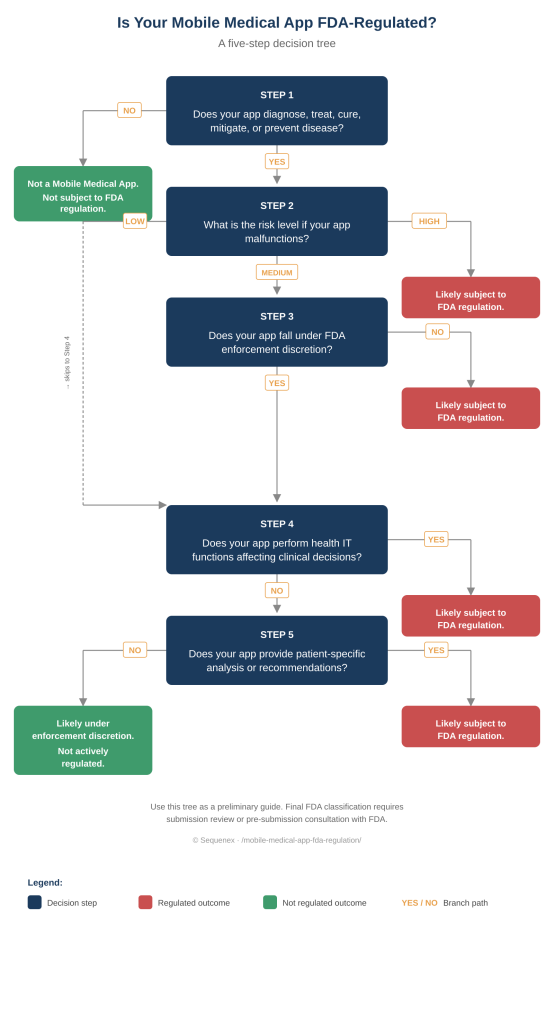
Step 1 — Does your app meet the definition of a medical device?
The FDA defines a medical device as any product intended to diagnose, treat, cure, mitigate, or prevent a disease or condition. If your app supports general wellness, fitness, or healthy lifestyle without making clinical claims — such as basic step counters, sleep tracking, or food logging — it does not meet the medical device definition and is not a mobile medical app under FDA regulation. If your app does meet the definition, continue to Step 2.
Step 2 — What is the risk level if your app malfunctions?
FDA risk classification considers what could happen if the app produces incorrect output, fails to operate, or is misused. Apps that could malfunction and cause serious injury or death (high risk—e.g., software driving insulin dosing decisions or interpreting cardiac arrhythmias) are very likely subject to FDA regulation. Apps with medium-risk implications continue to Step 3 for further evaluation. Apps with low-risk implications skip to Step 4.
Step 3 — Does your app fall under FDA enforcement discretion?
Enforcement discretion is the FDA’s policy of not actively enforcing regulatory requirements for certain low- to moderate-risk MMAs that technically meet the medical device definition but pose limited patient risk. The FDA’s published guidance on mobile medical applications lists categories that typically receive enforcement discretion — including apps that automate simple calculations, help patients self-manage chronic conditions without providing specific treatment recommendations, or organize and track health information. If your app fits one of these categories, continue to Step 4. If not, your app is likely subject to FDA regulation.
Step 4 — Does your app perform health IT functions affecting clinical decisions?
Some apps integrate with electronic health records (EHRs), share patient data across clinical systems, or perform health information technology (health IT) functions that influence clinical decision-making. Apps that present clinical recommendations to providers in ways that materially affect treatment choices — beyond simply transmitting or displaying data — are subject to FDA regulation. Apps that move data without interpreting it for clinical decisions, or that surface information in ways that leave clinical judgment fully with the clinician, generally are not. If your app’s health IT functions affect clinical decisions, your app is likely subject to FDA regulation. Otherwise, continue to Step 5.
Step 5 — Does your app provide patient-specific analysis or recommendations?
Apps that analyze patient-specific data and generate recommendations, diagnoses, or treatment guidance are evaluated as medical devices. This includes algorithms that interpret CGM data to suggest insulin dosing, ML models that flag arrhythmias from wearable ECG data, and decision support tools that recommend medication adjustments based on patient inputs. If your app provides patient-specific analysis, it is likely subject to FDA regulation. If it does not — providing only general information without patient-specific interpretation — your app likely falls under enforcement discretion and is not actively regulated.
Regulated vs Unregulated MMAs — Examples
The decision tree above identifies which category your app falls into. The examples below show what each category looks like in practice. The line between regulated and unregulated isn’t always obvious from a product description — intended use, the specificity of the analysis, and the clinical decisions the app influences all matter.
Examples of Regulated Mobile Medical Apps
Apps that drive or directly inform clinical decisions are typically subject to FDA regulation. Examples include: insulin dose calculators that recommend specific dosing based on glucose readings; ECG analysis apps that interpret cardiac rhythms and flag arrhythmias; wearable cardiac event monitors with FDA-cleared diagnostic algorithms; AI-based image analysis apps for dermatology, radiology, or pathology; closed-loop or hybrid closed-loop diabetes management algorithms; and clinical decision support apps that provide treatment recommendations beyond what a clinician would derive from displayed data alone.
Examples of Mobile Medical Apps Under Enforcement Discretion
Apps that support patients or providers without driving clinical decisions typically fall under enforcement discretion or are entirely outside FDA regulation. Examples include: medication reminder apps that don’t calculate dosing; health and wellness logging apps that track diet, exercise, mood, or symptoms without providing clinical interpretation; apps that automate simple medical calculations a clinician would otherwise do by hand; apps that organize patient health information for self-management; basic fitness trackers; and educational apps that present medical information without personalizing it to a specific patient’s clinical situation.
2026 FDA Regulatory Updates Affecting Mobile Medical Apps
FDA expectations for mobile medical apps have evolved significantly since 2023. Three updates matter most for any MMA developer working through an FDA submission in 2026: cybersecurity premarket requirements under section 524B of the Federal Food, Drug, and Cosmetic Act (now enforceable for every applicable submission), the FDA’s finalized guidance on AI/ML-enabled device software functions, and the framework for Predetermined Change Control Plans (PCCPs) that allows planned model updates without resubmission. The three updates work together — most modern MMAs need to address all three.
Cybersecurity Premarket Requirements (Section 524B)
Section 524B of the Federal Food, Drug, and Cosmetic Act, added through the Consolidated Appropriations Act of 2023, requires that FDA premarket submissions for cyber devices include comprehensive cybersecurity documentation. A cyber device is broadly defined as any device that contains software validated, installed, or authorized by the sponsor, thereby capturing the vast majority of mobile medical apps.
For mobile medical apps subject to FDA regulation, the cybersecurity submission package must include four components: a Software Bill of Materials (SBOM) inventorying every software component and its known vulnerabilities; threat modeling that documents potential attack vectors and mitigations using frameworks such as STRIDE or AAMI TIR57; vulnerability assessment evaluating known vulnerabilities in the app and its components, with remediation plans; and a post-market vulnerability monitoring plan defining how the sponsor will track, disclose, patch, and report security issues after market entry.
Apps that fail to include adequate cybersecurity documentation under section 524B face Refusal-to-Accept (RTA) decisions — meaning the FDA refuses to even begin substantive review of the submission until the cybersecurity package is complete. RTA decisions add weeks or months to clearance timelines. Building cybersecurity into the architecture from sprint zero, rather than retrofitting it before submission, is the practical way to avoid RTA delays.
AI/ML-Enabled Mobile Medical Apps
Mobile medical apps incorporating artificial intelligence or machine learning face additional regulatory scrutiny. The FDA’s evolving guidance on AI/ML-enabled device software functions covers expectations across the model lifecycle — from training data curation to post-market monitoring — and is particularly relevant for MMAs that use machine learning for clinical decision support, diagnostic interpretation, or predictive analytics.
Five expectations apply to most AI/ML-enabled MMAs. Training data must be characterized — including provenance, demographic distribution, clinical setting, and known biases. Model performance must be validated not just in aggregate but across clinically relevant subpopulations, including age, sex, race, ethnicity, and disease severity. The submission must explain algorithmically how the model produces outputs at a level appropriate to the device’s risk class. Human factors and interface design must be evaluated, especially for AI-driven decision-support apps, where output presentation can lead to over-reliance or misinterpretation. And bias evaluation must follow the FDA’s Good Machine Learning Practice (GMLP) guiding principles, published jointly with Health Canada and the UK MHRA.
AI/ML-enabled MMAs that update their models post-market — based on new training data, real-world performance feedback, or scheduled retraining — face an additional consideration: how to handle those updates without requiring a new FDA submission for every change. That’s where Predetermined Change Control Plans come in.
Predetermined Change Control Plans (PCCPs)
A Predetermined Change Control Plan (PCCP) is an FDA-aligned framework that allows a sponsor to define — in advance, at premarket submission — what modifications they intend to make to an AI/ML-enabled device after market authorization, and how those modifications will be implemented and validated. Modifications that fall within the PCCP’s defined scope can be deployed without a new FDA submission for each change. Modifications outside the PCCP’s scope still require new submissions.
PCCPs are particularly valuable for AI/ML-enabled MMAs because the value of these apps often increases with retraining on new data. Without a PCCP, every model update would trigger a new submission — a regulatory burden that effectively prevents iterative improvement. With a PCCP, planned updates within defined boundaries become a documented, repeatable process.
A complete PCCP includes a description of the modifications the sponsor intends to make, a modification protocol specifying how each change will be implemented and validated, and an impact assessment showing the modification falls within the PCCP’s intended scope and does not change the device’s intended use. PCCPs are increasingly common for AI/ML-enabled MMAs across diabetes care, cardiology, dermatology, mental health, and other therapeutic areas.
Frequently Asked Questions About Mobile Medical App FDA Regulation
What is a mobile medical app (MMA)?
A mobile medical app (MMA) is a software application running on a smartphone, tablet, or other mobile platform that performs a medical function — diagnosing, treating, curing, mitigating, or preventing disease. The FDA defines MMAs in its 2019 guidance on mobile medical applications and regulates those that meet the definition of a medical device and pose meaningful patient risk.
Is a fitness tracker considered a mobile medical app under FDA regulation?
Most fitness trackers are not considered mobile medical apps under FDA regulation. Apps that track steps, calories, sleep, or general wellness without diagnosing or treating disease fall outside the FDA’s definition of a medical device. However, fitness apps that include features like ECG analysis, blood pressure monitoring, or arrhythmia detection do meet the definition and are FDA-regulated — Apple Watch’s ECG feature is a well-known example.
What is FDA enforcement discretion for mobile medical apps?
FDA enforcement discretion is the agency’s policy of not actively enforcing regulatory requirements for certain low- to moderate-risk mobile medical apps that technically meet the medical device definition but pose limited patient risk. Apps under enforcement discretion can be marketed without 510(k) clearance, but they must still meet certain quality and labeling expectations. Enforcement discretion is not an exemption — the FDA can change its position if a product turns out to pose an unexpected risk.
What’s the difference between a Mobile Medical App and Software as a Medical Device (SaMD)?
Mobile Medical App (MMA) and Software as a Medical Device (SaMD) overlap heavily. SaMD is the IMDRF-defined term for software intended for medical purposes that performs those purposes without being part of a hardware medical device. MMA is the FDA’s term for the same category, specifically when the software runs on a mobile platform like a smartphone or tablet. Most MMAs are also SaMD; the terms are largely interchangeable, but SaMD is the broader and more international term.
Do mobile medical apps need to comply with FDA cybersecurity requirements?
Yes. Since 2023, FDA premarket submissions for mobile medical apps that address cybersecurity must include detailed cybersecurity documentation under section 524B of the Federal Food, Drug, and Cosmetic Act. Required content includes a Software Bill of Materials (SBOM), threat modeling, vulnerability assessment, and a plan for monitoring and addressing post-market vulnerabilities. Apps that fail to meet these requirements face refusal-to-accept (RTA) decisions, blocking their submission from substantive review.
How do AI and machine learning affect mobile medical app FDA regulation?
AI/ML-enabled mobile medical apps face additional scrutiny from the FDA. The FDA’s 2024 guidance on AI/ML-enabled device software functions covers expectations for training data, model validation, transparency, and bias evaluation. Apps with machine learning models intended to update post-market typically require a Predetermined Change Control Plan (PCCP) — an FDA-aligned framework that defines which model updates can occur without resubmission. PCCPs are increasingly common for AI-driven decision support, diagnostic, and predictive MMAs.
Determined Your App Is FDA-Regulated? Here’s What Comes Next.
If the decision tree confirmed your mobile medical app is subject to FDA regulation, the path from here typically runs through five steps: regulatory strategy and pathway selection (510(k), De Novo, or PMA), software lifecycle development under IEC 62304, quality management under ISO 13485, risk management under ISO 14971, and cybersecurity submission documentation under FDA section 524B.
Sequenex builds regulated mobile medical software every day, with all of these standards already operationalized inside an ISO 13485-certified QMS. Whether you’re navigating your first FDA submission or scaling an existing MMA, we can help map the regulatory path and execute the engineering work alongside it.